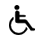 Pristupačnost
Pristupačnost
PROJEKT Adesire / PROJECT Adesire
IP-2016-06-3775![]()
Trajanje / Duration:
01.03.2017. - 30.06.2021.
Financiranje / Funding:
HRVATSKA ZAKLADA ZA ZNANOST / Croatian Science Fundation
VODITELJ PROJEKTA / PRINCIPAL INVESTIGATOR: prof. dr. sc. Tomica Hrenar
INSTITUCIJE KOJE SUDJELUJU U PROJEKTU / INSTITUTIONS
Sveučilište u Zagrebu Prirodoslovno-matematički fakultet, Zagreb / University of Zagreb Faculty of Science, Zagreb, Croatia
Sveučilište u Splitu Prirodoslovno-matematički fakultet, Split / University of Split Faculty of Science, Split, Croatia
NAZIV PROJEKTA / PROJECT TITLE
Aktivnošću i in silico usmjeren dizajn malih bioaktivnih molekula /
Activity and in silico guided design of bioactive small molecules
SAŽETAK / ABSTRACT
U ovom projektu predložena je sinteza, karakterizacija i biološka evaluacija novih tipova spojeva te ekstenzivni kvantno-kemijski proračuni i multi-way analize. Bioaktivni kemijski skeletni tipovi kao što su benzimidazoli, imidazoli i kinuklidini prepoznati su kao spojevi sa širokim spektrom bioloških aktivnosti, također i u području infektivnih bolesti te poremećaja središnjeg živčanog sustava (Alzheimerova bolest, AD). Mi predviđamo da bi daljnja istraživanja tih klasa kemijskih spojeva mogla rezultirati s identifikacijom novih spojeva prikladnih za daljnji razvoj u moguće lijekove. Kao jako inovativni dio ovog istraživanja, a za razvoj točnih kvantitativnih modela povezivanja strukture i reaktivnosti bit će korištene napredne metode molekularnog modeliranja u kombinaciji s multi-way analizama. Ti modeli će se iskoristiti za iterativni postupak usmjerenog racionalnog dizajna. U početku će se korištenjem klasičnih i suvremenih metoda organske kemije sintetizirati novi bioaktivni spojevi. Za priređene kiralne spojeve odredit će se stereoselektivnost njihovih interakcija. Za sve spojeve, odredit će se antimikrobna aktivnost osobito prema rezistentnim Gram-negativnim bakterijama. Nadalje, njihov potencijal za razvoj poboljšanih lijekova za AD odredit će se mjerenjem njihove inhibicije kolinesteraza. Za procjenu mogućih pozitivnih sinergijskih učinaka, sintetizirat će se hibridne molekule te će se testirati i smjese najaktivnijih sintetiziranih spojeva. Molekularno modeliranje, doking simulacije i novorazvijena metodologija za primjenu multi-way analiza na kombinirane eksperimentalne i teorijske podatke koristit će se za ciljane strukturne promjene koje bi mogle dovesti do novih i efikasnijih bioaktivnih skeletnih tipova s potencijalnom primjenom u biotehnologiji, farmaceutskoj industriji i medicini.
In this project synthesis, characterization, and biological evaluation of novel types of compounds including extensive quantum chemical calculations and multi-way analyses is proposed. Bioactive chemical scaffolds such as benzimidazoles, imidazoles, and quinuclidines are recognized as compounds with a broad spectrum of biological activities also in the fields of infectious diseases and central nervous system disorders (Alzheimer’s disease, AD). We envisioned that further exploration of these chemical classes can result with the identification of new leads suitable for further development into potential hits. As a highly innovative part of this study, advanced methods of molecular modelling combined with multi-way analysis will be used for construction of accurate quantitative structure–activity relationship models. These models will be used for iterative procedure of guided rational design. Initially, new bioactive compounds will be synthesized using classical and contemporary methods of organic chemistry. Chiral compounds will be prepared and the stereoselectivity of interactions evaluated. For all compounds, antimicrobial activity will be estimated targeting resistance genes to treat infections caused by multidrug-resistant Gram-negative bacteria. Furthermore, their perspective for development of enhanced anti-AD drugs will be assessed by determining their inhibitory potency toward cholinesterases. To evaluate possible beneficial synergistic effects, hybrid molecules as well as various combinations of the most active synthesized compounds will be prepared and tested. Molecular modelling, docking studies and a newly developed method for application of multi-way analysis to combined experimental and theoretical data will be used to direct structural changes leading to the synthesis of new, more efficient bioactive scaffolds with potential use in biotechnology, pharmaceutical industry and medicine.
PROJEKTNI TIM / PROJECT TEAM
Ines Primožič, Anita Bosak, Renata Odžak, Mirjana Skočibušić, Alma Ramić, Linda Bazina, Ricardo André Fernandes da Mata, Karlo Sović, Zlatan Spahić, Ana Mikelić
http://www.hrzz.hr/default.aspx?id=1205&pid=3775&rok=2016-06
PUBLIKACIJE PROIZAŠLE IZ PROJEKTA / PUBLICATIONS SUPPORTED BY THE PROJECT
12. Z. Spahić, T. Hrenar, I. Primožič: "Polytopal Rearrangement Governing Stereochemistry of Bicyclic Oxime Ether Synthesis", Int. J. Mol. Sci. 23 (2022) 12331.
IF2021=6.208
Q1(>10 %) in BIOCHEMISTRY & MOLECULAR BIOLOGY, Rank: 69/297
DOI: https://doi.org/10.3390/ijms232012331
11. A. Ramić, A. Matošević, B. Debanić, A. Mikelić, I. Primožič, A. Bosak, T. Hrenar: "Synthesis, biological evaluation and machine learning prediction model for fluorinated Cinchona alkaloid-based derivatives as cholinesterase inhibitors", Pharmaceuticals 15 (2022) 1214.
IF2021=5.215
Q1(>10 %) in PHARMACOLOGY & PHARMACY, Rank: 69/279
DOI: https://doi.org/10.3390/ph15101214
10. D. Raljević, J. Parlov Vuković, V. Smrečki, Lj. Marinić Pajc, P. Novak, T. Hrenar, T. Jednačak, L. Konjević, B. Pinević,
T. Gašparac: "Machine learning approach for predicting crude oil stability based on NMR spectroscopy", Fuel 305 (2021) 121561.
IF2021=8.035
Q1(>10 %) in ENGINEERING, CHEMICAL, Rank: 19/142
Q1(>10 %) in ENERGY & FUELS, Rank: 29/119
DOI: https://doi.org/10.1016/j.fuel.2021.121561
9. A. Ramić, M. Skočibušić, R. Odžak, A. Čipak Gašparović, L. Milković, A. Mikelić, K. Sović, I. Primožič, T. Hrenar: "Antimicrobial Activity of Quasi-Enantiomeric Cinchona Alkaloid Derivatives and Prediction Model Developed by Machine Learning", Antibiotics 19 (2021) 659.
Q2, IF2020=4.639 in PHARMACOLOGY & PHARMACY, Rank: 75/275
DOI: https://doi.org/10.3390/antibiotics10060659
8. A. Matošević, A. Radman Kastelic, A. Mikelić, A. Zandona, M. Katalinić, I. Primožič, A. Bosak, T. Hrenar: "Quinuclidine-Based Carbamates as Potential CNS Active Compounds", Pharmaceutics 13 (2021) 420.
Q1(>10 %), IF2020= 6.321 in PHARMACOLOGY & PHARMACY, Rank: 28/275
DOI: https://doi.org/10.3390/pharmaceutics13030420
7. A. Zandona, M. Katalinić, G. Šinko, A. Radman Kastelic, I. Primožič, Z. Kovarik: "Targeting organophosphorus compounds poisoning by novel quinuclidine‑3 oximes: development of butyrylcholinesterase‑based bioscavengers" Arch. Toxicol. 94 (2020) 3157–3171
Q1(>10 %), IF2020=5.153 in TOXICOLOGY, Rank: 16/93
DOI: https://doi.org/10.1007/s00204-020-02811-5
6. T. Jednačak, M. Majerić Elenkov, T. Hrenar, K. Sović, J. Parlov Vuković, P. Novak: "Solution and solid state studies of hydrogen bonding in substituted oxazolidinones by spectroscopic and quantum chemical methods", New J. Chem. 44 (2020) 6456–6463.
Q2, IF2020=3.591 in CHEMISTRY, MULTIDISCIPLINARY, Rank: 75/179
DOI: https://doi.org/10.1039/c9nj06349h
5. K. Sović, T. Ostojić, S. Cepić,A. Ramić, R. Odžak, M. Skočibušić, T. Hrenar, I. Primožič: "Conformational Analysis of Cinhonine and Cinhonidine by Tensor Decomposition of Molecular Dynamics Trajectories", Croat. Chem. Acta 92 (2019) 259–267.
Q4, IF2019=0.812 in CHEMISTRY, MULTIDISCIPLINARY, Rank: 151/177
DOI: https://doi.org/10.5562/cca3557
4. A. Radman Kastelic, R. Odžak, I. Pezdirc, K. Sović, T. Hrenar, A. Čipak Gašparović, M. Skočibušić, I. Primožič: "New and Potent Quinuclidine-Based Antimicrobial Agents", Molecules 24 (2019) 2675.
Q2, IF2019=3.267 in CHEMISTRY, MULTIDISCIPLINARY, Rank: 63/179
DOI: https://doi.org/10.3390/molecules24142675
3. T. Hrenar, I. Primožič, D. Fijan, M. Majerić Elenkov: "Conformational analysis of spiro-epoxides by principal component analysis of molecular dynamics trajectories", Phys. Chem. Chem. Phys. 19 (2017) 31706–31713.
Q1(>10 %), IF2017=3.906 in PHYSICS, ATOMIC, MOLECULAR & CHEMICAL, Rank: 9/37
DOI: https://doi.org/10.1039/c7cp05600a
2. A. Bosak, A. Ramić, T. Šmidlehner, T. Hrenar, I. Primožič, Z. Kovarik: "Design and evaluation of selective butyrylcholinesterase inhibitors based on Cinchona alkaloid scaffold", PLoS One 13 (2018) e0205193.
Q2, IF2018=2.776 in MULTIDISCIPLINARY SCIENCES, Rank 26/73
DOI: https://doi.org/10.1371/journal.pone.0205193
1. M. Skočibušić, R. Odžak, A. Ramić, T. Smolić, T. Hrenar, I. Primožič: "Novel Imidazole Aldoximes with Broad-Spectrum Antimicrobial Potency against Multidrug Resistant Gram-Negative Bacteria", Molecules 23 (2018) 1212.
Q2, IF2018=3.060 in CHEMISTRY, MULTIDISCIPLINARY, Rank 80/216
DOI: https://doi.org/10.3390/molecules23051212
KLJUČNI REZULTATI / KEY RESULTS
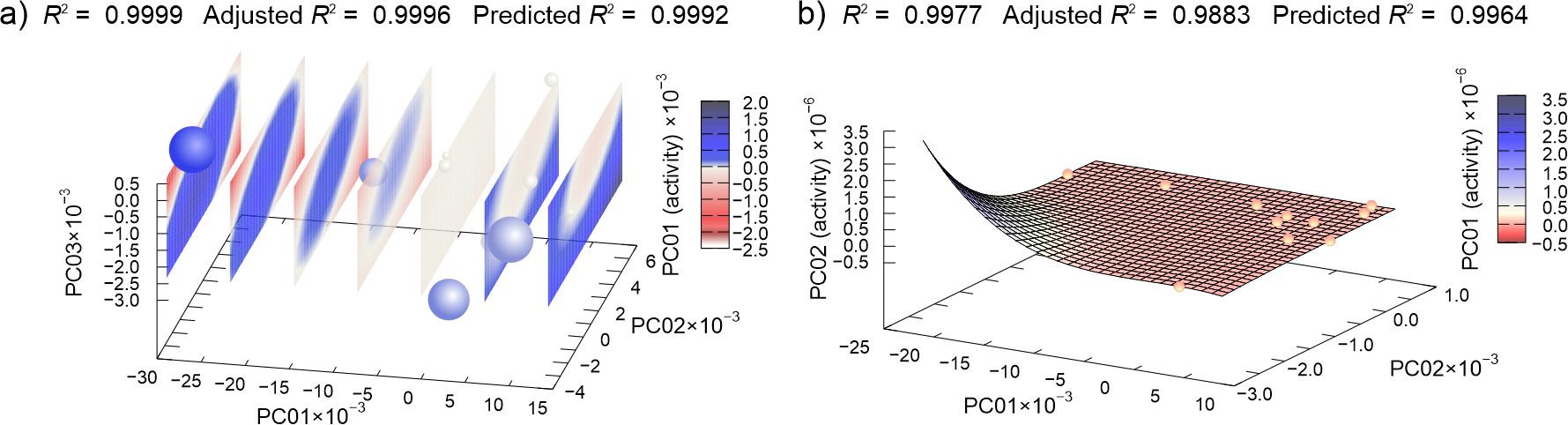
Figure 1. Machine learning determined best multivariate regression models of (a) cinchonidine and (b) cinchonine MIC data dependent on the principal component of compounds potential energy surfaces. (In (a), spheres represent points in 3D-reduced space, and the planes are cuts of polynomial regression model; for easier interpretation, the fourth dimension is represented redundantly with the color and size of the spheres.)
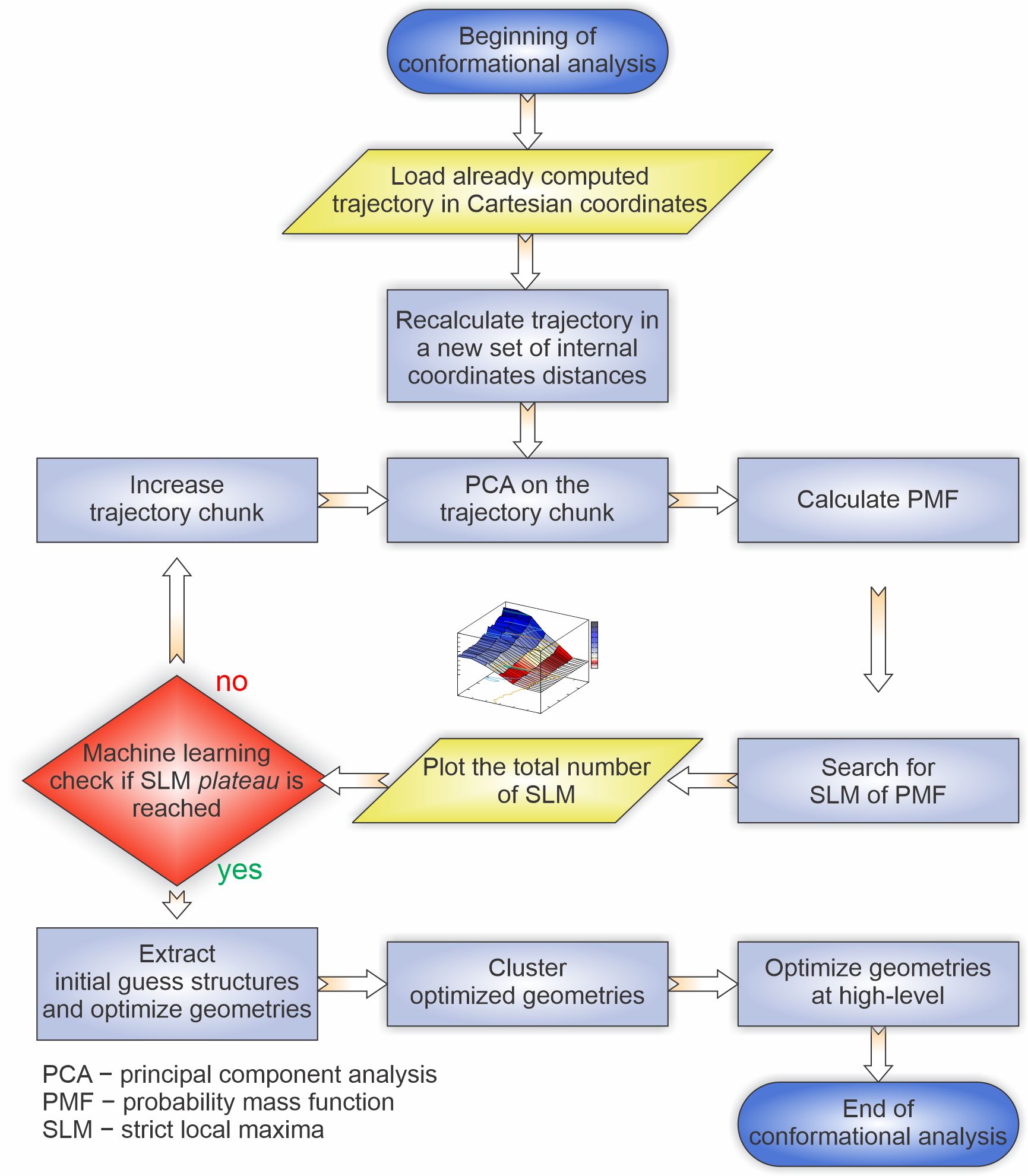
Figure 2. Machine learning protocol for full conformational analysis from tensor decomposition of molecular dynamics trajectories using the strict local maxima plateau criteria.
Figure 3. Probability distribution of molecular geometries in the reduced space calculated by principal component analysis of the molecular dynamics trajectory. Strict local maxima plateau is reached at 1000000 points and 3 principal components.
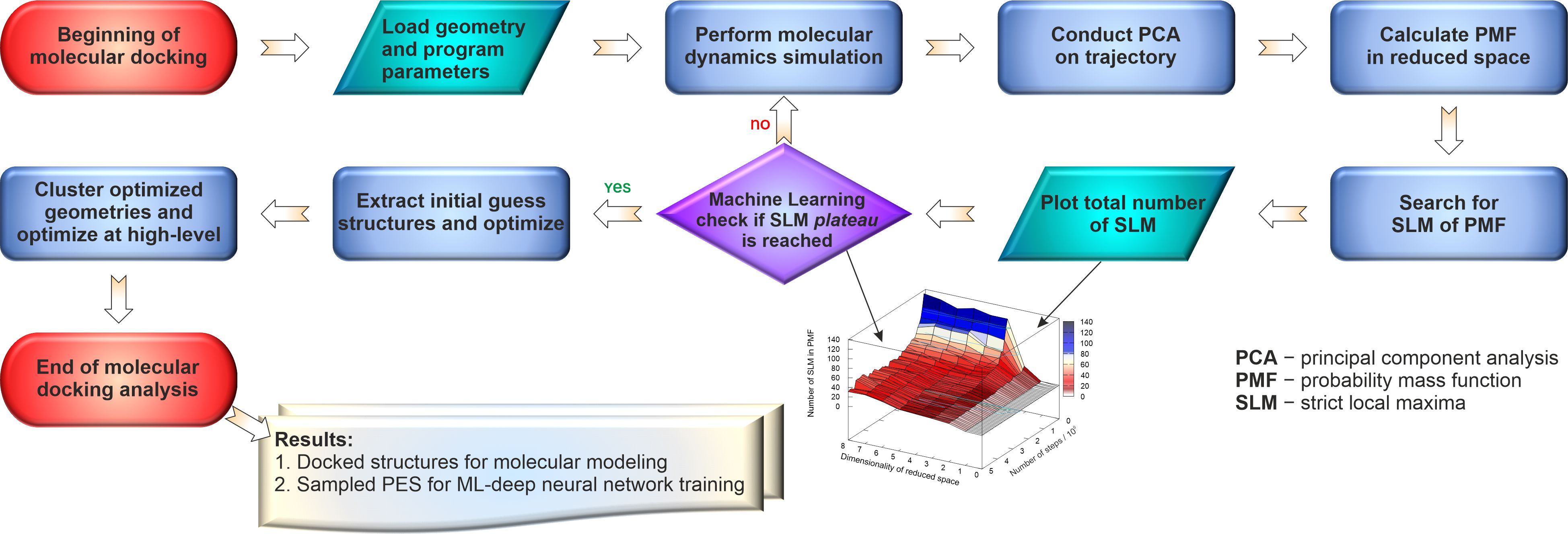
Figure 4. Machine learning protocol for ab initio molecular docking from tensor decomposition of molecular dynamics trajectories using the strict local maxima plateau criteria.
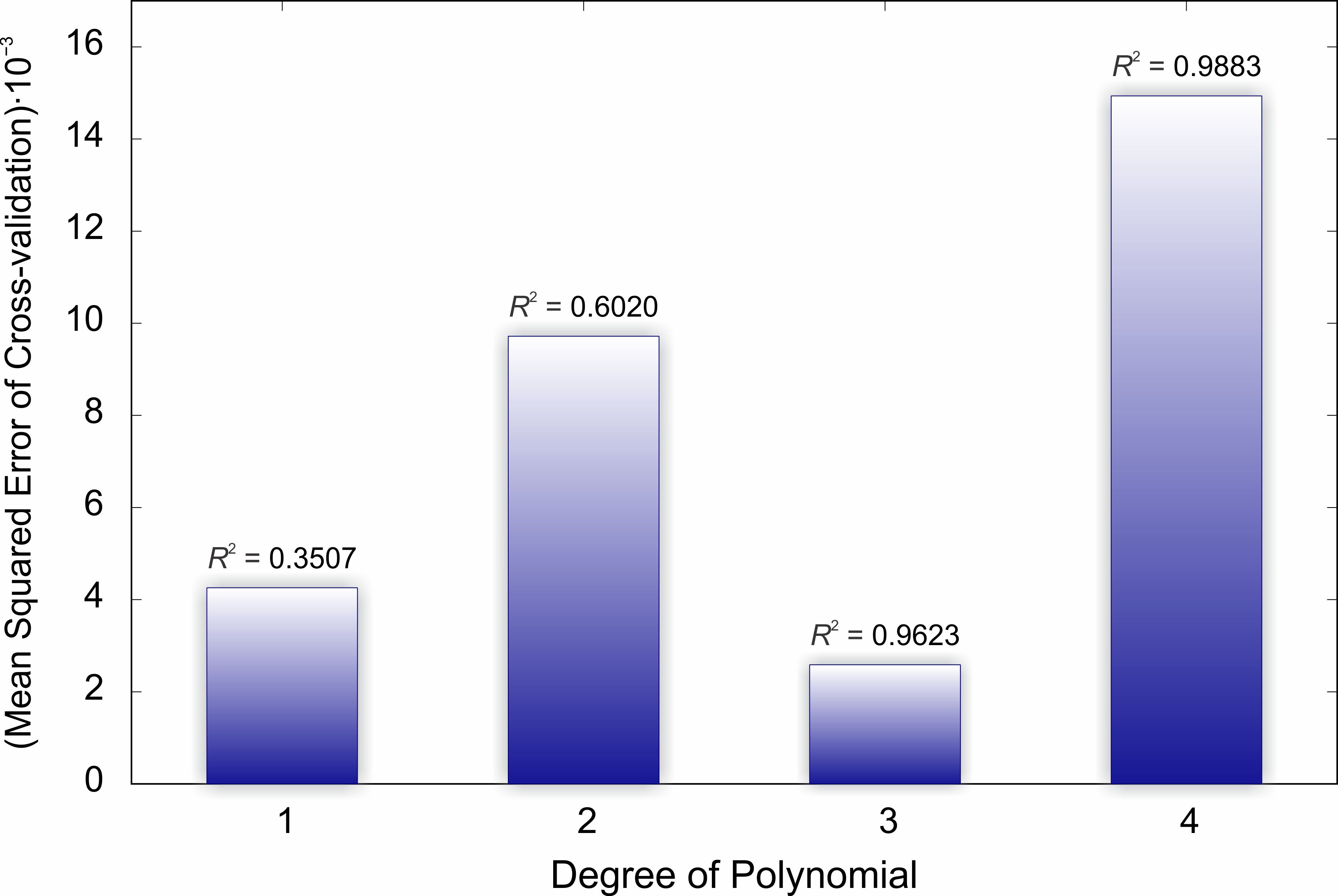
Figure 5. Leave-one-out cross validation for PLS regression of potential energy surfaces to biological activity data.